The Science Behind COVID-19
This post originally appeared on Dr. Vincent Cannataro’s personal blog. Updated resources and additional links may be available there as COVID-19 progresses.
I spent a large chunk of my graduate training researching and modeling novel pathogen spread, and I am currently teaching both a “Current Topics in Biological Research” class and an “Introduction of Evolutionary Biology” class (wherein we discuss viral genomes and phylogenetic relationships)… so I have been thinking and teaching about COVID19 all semester (which just about coincides with the full timeline of COVID19 discovery and spread). Thus far most of my professing has been largely ephemeral—a slide here and there to relate some science to current real-world research. In this post, I hope to create a more permanent and updatable container of what we know and how we know it. I am not a virologist or an epidemiologist, so I will just write about general science here, and refer readers to “the experts” at the end of the post.
How can scientists infer the trajectory of viral spread?
Viruses are relatively simple. Typically just a few genes wrapped up in an envelope of proteins and fats (aside: this is why hand washing with soap is effective against COVID19. Soap breaks up fats—like the COVID19 envelope—and thus effectively destroys the virus). Scientists can harness the relative simplicity of a specific virus, that virus’ genome, and the virus’ mechanism and means of transmission, to make strong predictions of where that virus has been, and where that virus may go.
Where the virus has been
How can we tell where a virus came from? I mentioned that each virus has its own “genome”—that is, a set of genes that control the various viral mechanisms. These genes get replicated every time the virus itself replicates. Now, if you have read my blog before, you know that I like to stress that gene replication is an imperfect process. The imperfect nature of DNA replication means that viral genes can accumulate differences from one another as the virus spreads among people. We can use these genomic differences—physical differences in the exact nucleic acids that constitute the viral genome—to build a tree of relatedness among viruses that had their genome sequenced.
The tree of relatedness (called a phylogenetic tree in evolutionary biology) demonstrates the historical interconnectedness of the viruses because mutations happen randomly, and accumulate one-after-the-other, meaning that if virus A and virus B share the same difference at the same location when compared to all other viruses, then A and B are likely descended from a relatively recent common ancestor. And if a set of viruses from location Y are newly sequenced, and found to all share the same genomic differences with a set of viruses sequenced last week from location X, then you can bet that the Y viruses are related to (and, maybe even potentially originated from) the X viruses.
You can explore a continually-updated phylogenetic tree of COVID19 over at https://nextstrain.org/ncov.
So, we can reconstruct the history of spread of a virus by pinning down the relatedness of viral genomes in space and through time. But, how can we predict where the virus will go?
Where the virus is going
Again, we can rely on the relatively simple nature of the virus itself to make predictions about viral dynamics. By watching how readily and quickly the pathogen originally spreads throughout a community epidemiologists can calculate an average “basic reproduction number” that tells us, essentially, how many new infections may arise from a single current infection (without any sort of intervention). If this number is less than one, it means that on average each new case gives rise to less than one new case, and infectious agents die out. If the number is greater than one, each new case gives rise to more than one new case, and infections may spread.
One of the main goals of epidemiologists and public health officials during an epidemic is to get the reproduction number of a pathogen (sometimes now called the effective reproductive number because it is in the presence of intervention) down below 1 using various means of action. A major successful intervention for many viruses is vaccination, whereby you remove individuals from the “susceptible” pool and add them to the “resistant” pool, and effectively stop the spread of a pathogen in its tracks (one new infection cannot give rise to other infections if everyone who contacts the infected person is already immune!)
But, vaccination only works if the virus has been around for a while and scientists have had time to develop and test the efficacy of vaccines. What can we do about a novel virus, with no current vaccine?
One thing we can do, when we detect someone who tests positive for the virus, is track contacts of infected individuals and then isolate individuals that may be infectious. For instance, using a wide range of parameters initially estimated for COVID19, outbreaks in new areas were simulated to be stopped if a large percent of contacts of infectious individuals are both traced and isolated.
What if there is very little testing for the virus, and thus no way to trace newly infected individuals and their contacts? How can we slow the spread of a virus if we do not know exactly who has the virus? Other mechanisms, such as “social distancing”, also reduce the reproductive number of pathogens by changing human behavior to minimize the probability of both contracting and spreading a pathogen.
Why are scientists and health care experts advocating “social distancing?”
Imagine two scenarios:
- A major city holds a large parade, millions of people show up from around the region, all standing shoulder-to-shoulder for hours.
- A major city detects the presence of a novel virus at very small numbers within its population. Public health officials recommend social distancing, and thus parades and other large gatherings are cancelled.
It is easy to imagine how scenario (1) above would much more readily facilitate the transmission of a virus compared to scenario (2).
Once the spread of a new virus has begun in a densely populated area, slowing the spread of the virus is of paramount public health importance. Hospitals have limited capacity and medical resources are finite, so slowing the incidence of newly infected individuals maximizes the chance that hospital beds are available at any point in time, whereas rapid transmission of a virus can put all the strain of an epidemic on the healthcare system at the same time. This concept of “lowering the epidemic peak” is illustrated nicely in the figure below.
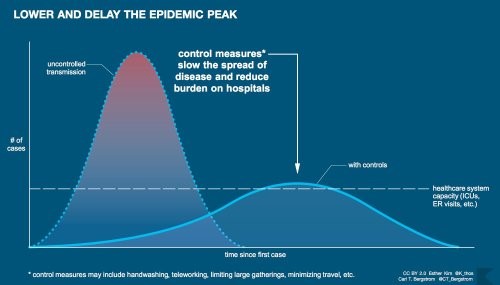
The example of the parade I used above came from real life. During the 1918 flu season Philly hosted a parade that resulted in the rapid spread of the flu. Other cities practiced social distancing once they learned of the infection, and had better public health outcomes. You can see how St. Louis “flattened the curve” here.
Data cleaning, analyses, and plots
If you are interested in the COVID19 case data, the team at Johns Hopkins University Center for Systems Science and Engineering has been curating and posting data here. I have a public github repository here, where I clean and do some simple analyses on this data, and plot a set of figures to track confirmed cases. Some outputs are also collated in a markdown document.
Links to the experts
The points below are by no means exhaustive, and I encourage readers to suggest more resources! These are resources I have found useful while I stumble around the web and encounter COVID science.
- Dr. Richard Lenski has a great blog, here. Dr. Lenski has been cataloging what we know and how we know it throughout the spread of COVID19, and I highly recommend you head over there for lots more information.
- Dr. Trevor Bedford has been tweeting and blogging about their and their colleagues’ analyses of the COVID19 genome and associated phylogenetics (updated live). Their amazing work was the first example (I personally saw) that demonstrated there may be cryptic transmission of the virus within the USA.
- Dr. C.T. Bergstrom has an informative Twitter feed, actively counters misinformation, and has created open-source material about COVID19.
- The CDC has information on preventing virus transmission at home and within communities.
- The Washington Post has a great simulation of an “SIR model” that illuminates how and why social distancing is effective in slowing viral spread. Find the simulation here.
- Johns Hopkins University coronavirus resource center has resources for both understanding COVID19 and also tracking the spread of the virus.
- This interactive R shiny app lets you experiment with disease parameters and interventions.


 About Emmanuel
About Emmanuel